

Sci. Pharm. 2024, 92(2), 23; https://doi.org/10.3390/scipharm92020023 - 25 Apr 2024
Abstract
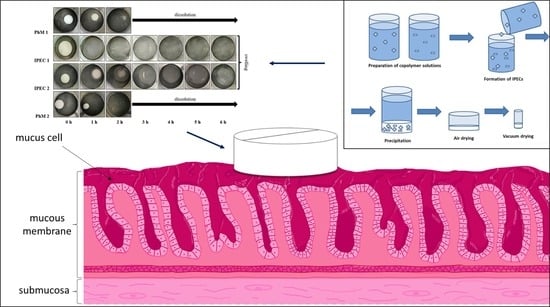
Hyaluronic acid (HA), as one of the main components of the extracellular matrix (ECM), plays an important role in the process of wound-healing and tissue-repair processes due to its unique properties and different physiological functions. HA has an ability to maintain a moist
[...] Read more.
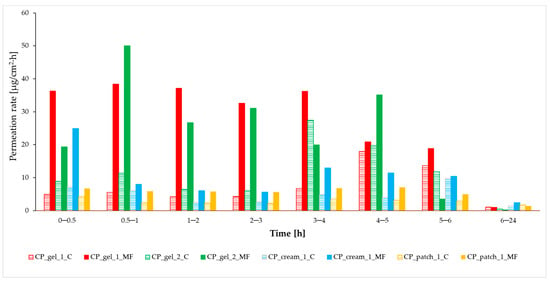
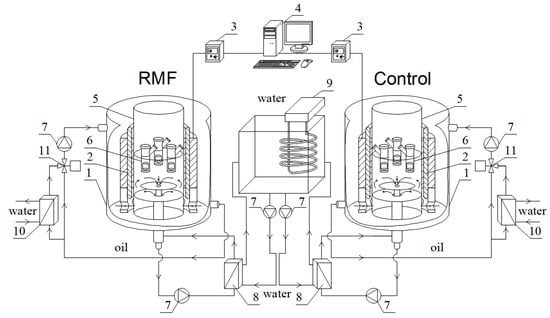
Hyaluronic acid (HA), as one of the main components of the extracellular matrix (ECM), plays an important role in the process of wound-healing and tissue-repair processes due to its unique properties and different physiological functions. HA has an ability to maintain a moist environment that promotes healing, the stimulation of growth factors and cellular constituents, and the migration of various cells essential for healing. This paper offers a review of HA use in the process of wound healing, with emphasis on hard-to-heal wounds, and examines its various applications in ophthalmology and otorhinolaryngology. It proves HA to be a versatile agent which finds its use in various fields of medicine for its antioxidant, anti-inflammatory, antibacterial properties and accelerated wound healing.
Full article
(This article belongs to the Special Issue Feature Papers in Scientia Pharmaceutica)



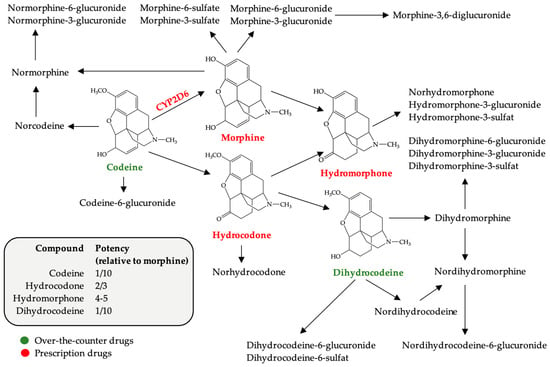
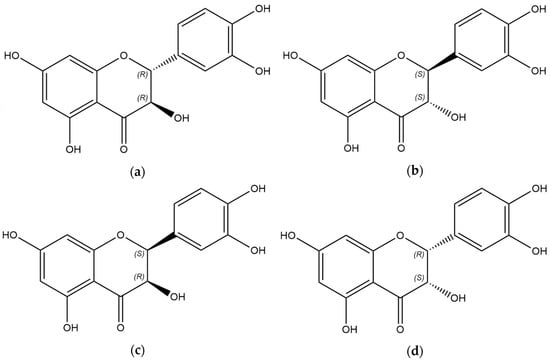
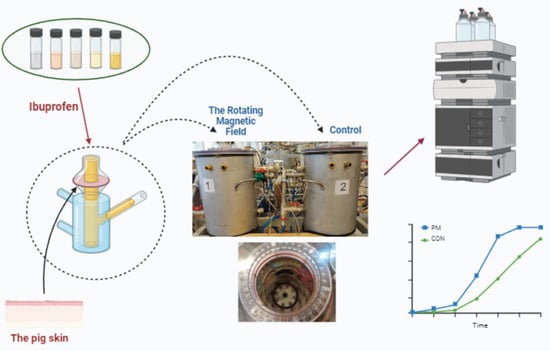

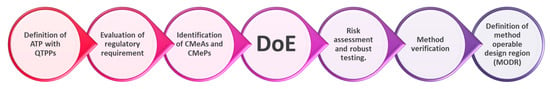{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}